Over 40 years in protein expression and purification – a historical perspective
by Christopher H. Taron, Ph.D., James C. Samuelson, Ph.D., and Lydia Morrison, M.S., New England Biolabs, Inc.
New England Biolabs® (NEB®) has been integrally involved in expressing and purifying proteins since the dawn of the recombinant DNA era in the 1970s – whether it be for our own research interests for our manufacturing processes. In 1978, NEB began screening microorganisms for restriction enzymes. Our scientists remember the challenges involved in purifying limited amounts of restriction enzymes and other proteins from these native organisms isolated from the environment. The efforts of NEB scientists to clone, overexpress and purify restriction enzymes from recombinant systems greatly advanced the field of molecular biology. Many of the original methods used by NEB scientists have endured and have been applied by countless scientists to study the structure and function of individual proteins. Now NEB scientists are striving to develop faster, simplified methods for recombinant protein expression and purification which rely on engineered protein expression hosts or optimized cell-free systems.
The period from 1966-77 saw a series of remarkable scientific breakthroughs. During this time, the genetic code was correctly interpreted, the first gene was isolated, and enzymes that both cut DNA at specific sequences (restriction enzymes) and that paste DNA pieces together (DNA ligases) were discovered. These discoveries ultimately enabled the cloning of the first genes and the creation of the first genetically modified microorganisms. Finally, in 1977, DNA sequencing technologies advanced beyond the laborious extension of just a few bases at a time and gave scientists the ability to unlock the genetic information encoded in any piece of DNA. The remarkable scientific advances of this decade, which made possible protein overexpression and purification, forever changed the course of biological and medical research, and enabled the emergence of the biotech industry (Figure 1).
NEB was founded in the midst of this era (1974) with the goal of providing researchers purified restriction enzymes, DNA ligases and other tools needed to clone and express genes. Restriction enzymes were the cornerstone of our early product offering. At that time, restriction enzymes were purified from bacteria isolated from the environment. This presented many challenges for commercial-scale production. For example, native restriction enzymes are generally not abundantly expressed and must be purified free from many other nucleases produced by an organism. Additionally, there were difficulties associated with large-scale culturing of various obscure microorganisms. Thus, to meet a steadily growing demand for these molecular tools, and to lower costs for our customers, NEB turned to recombinant DNA technology to clone and express enzymes in the laboratory bacterium, Escherichia coli (E. coli). This effort resulted in NEB producing some of the first recombinant enzymes available for commercial sale, and was the beginning of NEB’s long-tenured experience with the process of recombinant protein expression. Since these early days, recombinant protein expression has been integral to the success of NEB. Over the past forty years, we have continuously worked to invent and adopt new expression methodologies to improve the production of recombinant proteins. Our expertise has enabled the commercialization of over 550 recombinant enzymes to date. In this article, we highlight some of the major innovations in protein expression that have impacted our company’s journey, with both a historical view and an eye to the future.
Figure 1: Advances in DNA Understanding were Foundational for Protein Overexpression*
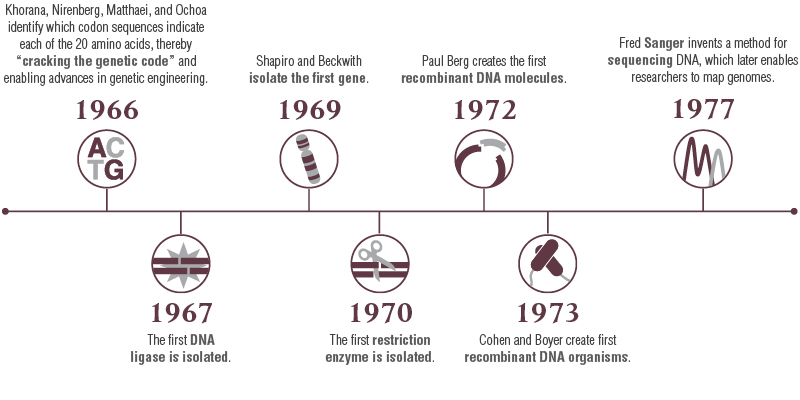
*Created referencing the National Science Teaching Association’s “Cloning Timeline”.
Early recombinant protein expression in E. coli
NEB’s interest in recombinant proteins was clearly evident by 1980. That year, the first recombinant enzymes were offered for sale. These enzymes were E. coli DNA Polymerase (Pol I), which was cloned by Bill Kelly in Noreen Murray’s lab at Edinburgh University, and T4 DNA Ligase, cloned by Geoff Wilson in the same lab several years earlier. Dedicated research on protein expression at NEB also commenced that year – including efforts to create a vaccine against malaria using recombinant parasite surface antigens. The cloning and expression methodology being used was quickly adopted for use with restriction enzymes to increase yields, enable higher purity, and permit better characterization of restriction enzyme structure and function. NEB’s early work involved establishing methods and tools to enable restriction enzyme cloning in E. coli, which had already become the standard for cloning and expression, and remains so today (1). In order to clone foreign restriction-modification systems in E. coli and over-produce individual restriction enzymes, it was necessary to characterize and eliminate the native methyl-dependent restriction systems of E. coli. Many of the key relevant discoveries were made by NEB scientists, who then genetically-tailored E. coli strains to be tolerant of restriction enzymes (2).
Cloning Vectors and Promoters
NEB’s first efforts in cloning used the E. coli plasmid pBR322, an early plasmid vector made by Francisco Bolivar and Ray Rodriguez, who were post-docs in Herb Boyer’s lab at the University of California, San Francisco. Incidentally, it was Herb Boyer who discovered EcoRI and demonstrated that the “sticky” ends it created could join DNA fragments from different sources, making it the first restriction enzyme useful for DNA cloning. NEB used derivatives of pBR322 that carried λPL, a powerful leftward promoter from bacteriophage Lambda, which is controlled by temperature (“off” at 32°C and “on” at 42°C). As pBR322 had only a moderate copy number (~30-40 copies per cell), NEB quickly adopted use of the higher copy number plasmid, pUC19, after its development by Jo Messing at the University of California Davis. The pUC19 vector offered multiple cloning sites, a much higher copy number (~250 copies per cell) and employed a promoter from the lac operon. In 1984, William Studier of Brookhaven National Labs developed an inducible T7 promoter system. With this method, a target gene is cloned downstream of the T7 promoter that is recognized by T7 RNA Polymerase (whose gene is integrated into the E. coli genome in expression strains). This strong promoter system is often capable of producing heterologous proteins, comprising up to 50% of total cellular protein. This approach became popular both at NEB and throughout the field.
NEB’s internal efforts on recombinant restriction enzymes soon paid off. In 1982, PstI became the first product cloned and expressed by NEB scientists. The recombinant strain overexpressed PstI ~100- fold relative to the native organism. This allowed NEB to reduce the unit price of PstI 20-fold (i.e., supplying 20 times more enzyme for the same price). Following PstI, NEB cloned, overexpressed and sold an increasing number of restriction enzymes each year, beginning with EcoRI, HaeII, HindIII, followed by many more. Today, nearly all of the over 250 restriction enzymes we sell are purified from overexpression clones made at NEB.
Purification using Affinity Chromatography
Soon after NEB began producing recombinant restriction enzymes, there was a desire to couple more facile purification to the expression process. In the mid-1980’s NEB began research on one of the first affinity-tagging systems. This approach employed fusing the gene encoding the E. coli maltose binding protein (MBP) in-frame with the target gene of interest. The resulting “fusion” protein can then be purified on amylose chromatography resin and the fusion tag can be removed using a site-specific protease. This system (the pMAL™ Protein Fusion and Purification System) was released in 1988 and was NEB’s first kit that enabled customers to perform protein expression and purification with the same system. As an added benefit, it was later discovered that MBP has the natural ability to significantly increase the solubility of fused target proteins in E. coli.
In the following years, the interest around affinity tags exploded. Additional fusion proteins (e.g., glutathione S-transferase [GST], chitin binding domain [CBD]) and many small peptide tags (poly-His-, FLAG-, S-tag-, Strep II- and poly-Arg-) were developed and used. Of these, the most influential was poly-His-tagging, which was developed by Roche in the late 1980’s. His-tagged fusion proteins can be recovered using immobilized metal affinity chromatography (IMAC), which typically employs Ni2+ beads or resin. To the present day, poly-His-tagged protein expression and IMAC is the most common approach to affinity-based protein purification, as it tolerates a wide range of conditions, including the presence of protein denaturants, high salt and detergents. It can also be used with many common cell lysis reagents and a variety of buffer additives.
The removal of an affinity tag/fusion partner from a purified recombinant protein is commonly performed using digestion with site-specific proteases. A drawback to this approach is that the released target protein needs to be purified from the liberated tag and the protease through additional chromatography steps. If the fusion partner contains the same affinity tag as the protease, this simplifies purification of the target protein. An increasingly popular approach is to remove both the fusion partner (e.g., 6His-MBP) and the protease (His-tagged TEV) by a single IMAC capture step. This technique is employed in the NEBExpress MBP Fusion and Purification System (Figure 2).
Figure 2: Overview of the NEBExpress™ MBP Fusion and Purification System (previously known as the pMAL Protein Fusion and Purification System)
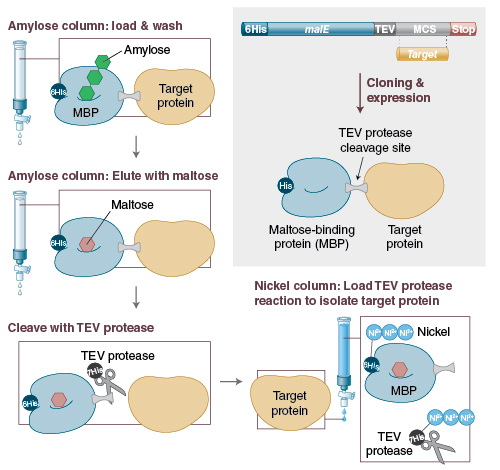
The target protein is fused to MBP, enhancing solubility and expression, which is followed by an easy and effective purification strategy.
Another NEB approach to affinity protein purification involved the use of auto-splicing protein domains called “inteins”. An intein was first described in 1988 in the context of protein splicing. In 1990, the first proof was provided that defined an intein as a protein domain that can catalyze its own excision from a protein. NEB researchers were studying inteins due to their presence in certain hyperthermophilic DNA polymerases, and described the intein reaction mechanism. Soon after, this research converged with protein expression and resulted in a new intein-mediated strategy for fusion protein removal without the need for protease cleavage. In this approach, E. coli expression of a target protein carrying an intein-chitin binding domain (intein- CBD) tag permits one-step purification using chitin resin. Upon passage of a cell lysate over chitin resin, the fusion protein becomes immobilized, after which the target protein can be released from CBD by inducing intein auto-cleavage with addition of a thiol-containing buffer or by pH shift. This work was commercialized as NEB’s IMPACT™ (Intein Mediated Purification with an Affinity Chitin-binding Tag) Kit in the late-1990s (3).
Figure 3: Expression of protein with multiple disulfide bonds using SHuffle® Competent E. coli
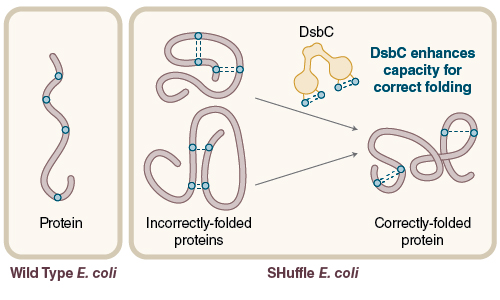
Disulfide bond formation in the cytoplasm of wild type E. coli is not favorable, while SHuffle is capable of correctly folding proteins with multiple disulfide bonds in the cytoplasm.
Solving Protein Expression Problems
As NEB has grown, so has our need to express classes of proteins outside of restriction enzymes. This has presented new challenges – as not all proteins express well, or at all, in E. coli. In addition to offering the popular BL21 and BL21(DE3) expression strains, NEB has focused on solving expression of “difficult” proteins. We have sought to improve the ability of E. coli to express various challenging proteins, including those with multiple disulfide bonds, with transmembrane domains, or that are toxic to the host.
Expressing Proteins Containing Disulfide Bonds
Disulfide bonds are post-translational covalent linkages formed by the oxidation of a pair of cysteines. Native disulfide bonds increase the stability of a protein and are often found in proteins that reside outside the chaperone rich environment of the cytoplasm, such as secreted peptides, hormones, antibodies, interferons and extracellular enzymes. When proteins are expressed in E. coli, it can be difficult for them to fold correctly. In 2009, NEB commercialized SHuffle® expression strains, which are engineered to support correct folding of proteins with multiple disulfide bonds in the cytoplasm (Figure 3). These strains constitutively express DsbC disulfide isomerase within the cytoplasm to promote the correction of mis-oxidized proteins (4).
Membrane or Toxic Protein Expression
Expression of membrane proteins is challenging for most heterologous systems, and often results in protein aggregation and misfolding due to the hydrophobic nature of transmembrane segments. When working with E. coli as a host, it is advantageous to express membrane proteins in moderation to avoid saturation of the membrane protein biogenesis pathway. NEB’s Lemo21(DE3) Competent E. coli strain was designed for tunable protein expression to achieve optimal assembly of transmembrane proteins or the optimal folding of soluble proteins (Figure 4) (5).
In cases where the heterologous protein is toxic to cells, tightly controlling gene expression can improve host viability by maintaining expression levels of a toxic target protein just below a host strain's tolerance. In strong T7 promoter-based systems, an effective means to control expression is to employ a host strain that expresses a T7 RNA Polymerase inhibitor protein (LysY) as in NEB’s Lemo21(DE3) or T7 Express lysY/Iq strains.
To express a highly toxic protein, it may be necessary to employ a cell-free expression system: NEB’s PURExpress® In Vitro Protein Synthesis Kit is reconstituted from purified components necessary for E. coli translation. This kit can also be used with the PURExpress Disulfide Bond Enhancer to improve protein folding. Alternatively, the NEBExpress™ Cell-free E. coli Protein Synthesis System utilizes a cell lysate which provides high-level expression of target proteins from linear or plasmid DNA templates.
Figure 4: Western analysis of 6-His tagged Brugia malayi protein
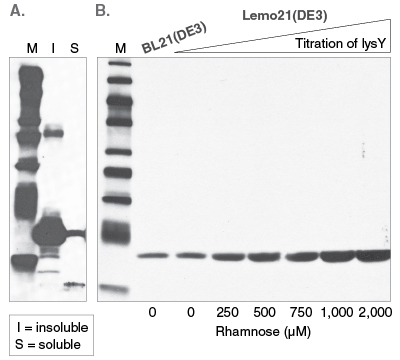
A) B. malayi protein expressed at 20°C in BL21(DE3).
B) Soluble fractions of B. malayi protein expressed at 30°C in BL21(DE3) or Lemo21(DE3).
The Future of Protein Expression
The protein expression field is constantly evolving. Applications such as protein engineering and synthetic biology are driving the field toward high throughput protein expression. Scientists now desire to test hundreds, if not thousands, of expressed proteins in a single day to quickly narrow their focus to the most interesting variants. As the standard method of cloning, vector introduction into a host strain, and cell propagation takes multiple days, it is becoming clear that cell-free protein expression, which can be accomplished in as little as one hour, will become increasingly important in the coming years. Just as in vivo protein expression started from humble beginnings and has progressed to highly engineered host strains and regimented bioprocessing, we anticipate a similar revolution in cell-free protein expression systems. A new generation of NEB scientists are dedicated to advancing cell-free expression by engineering novel cell lines, developing improved cell-free system manufacturing processes (such as those employed for PURExpress or NEBExpress), optimizing cell-free system formulations and exploring the potential for system scale up for production of milligram to gram quantities of protein.
View a PDF of this feature article
References
1. Rosano, G.L. and Ceccarelli, E.A. (2014) Front. Microbio. 5, 172.
2. Raleigh, E.A. and Wilson, G. (1986) PNAS, 83, 9070–9074.
3. Chong, S. et al. (1997) Gene, 192, 271-281.
4. Lobstein, J. et al. (2012) Microb. Cell. Fact. 11, 56.
5. Wagner, S. et al. (2008) PNAS, 105, 14371-14376.

